Treating and Managing Asthma
The long-term goals of asthma treatment are symptom control and risk reduction of exacerbations and asthma-related mortality.1 To achieve these goals, treatment includes inhaled corticosteroid (ICS)-containing medicine and a reliever inhaler for all asthma patients, treating modifiable risk factors, and optimizing non-pharmacologic strategies, including inhaler technique, regardless of the type of asthma.1
Mild to moderate asthma management
The preferred option in adults and adolescents is low-dose ICS (budesonide/beclometasone-formoterol) for both maintenance and reliever treatment (MART)1–3:
- For MART, the usual dose of budesonide/formoterol is 200/6 µg metered dose (160/4.5 µg delivered dose) and the usual dose of beclomethasone/formoterol is 100/6 metered dose (87.5/5 µg). Each of these combinations are prescribed as one inhalation twice daily and one inhalation as needed for symptom relief
- ICS/formoterol should not be used as the reliever for patients taking a different ICS/LABA maintenance treatment
Randomized clinical trials have provided evidence that in adolescents and adults with asthma, ICS/formoterol as reliever therapy reduces the risk of severe exacerbations compared with SABA reliever therapy, across the spectrum of asthma severity, when the same baseline maintenance treatment is taken (see table).1

Difficult to treat and moderate to severe asthma management

After confirming that symptoms are asthma-related, next steps include optimizing management. This can be done by evaluating for modifiable factors that could be contributing to uncontrolled symptoms and exacerbations and implementing strategies to overcome them.3
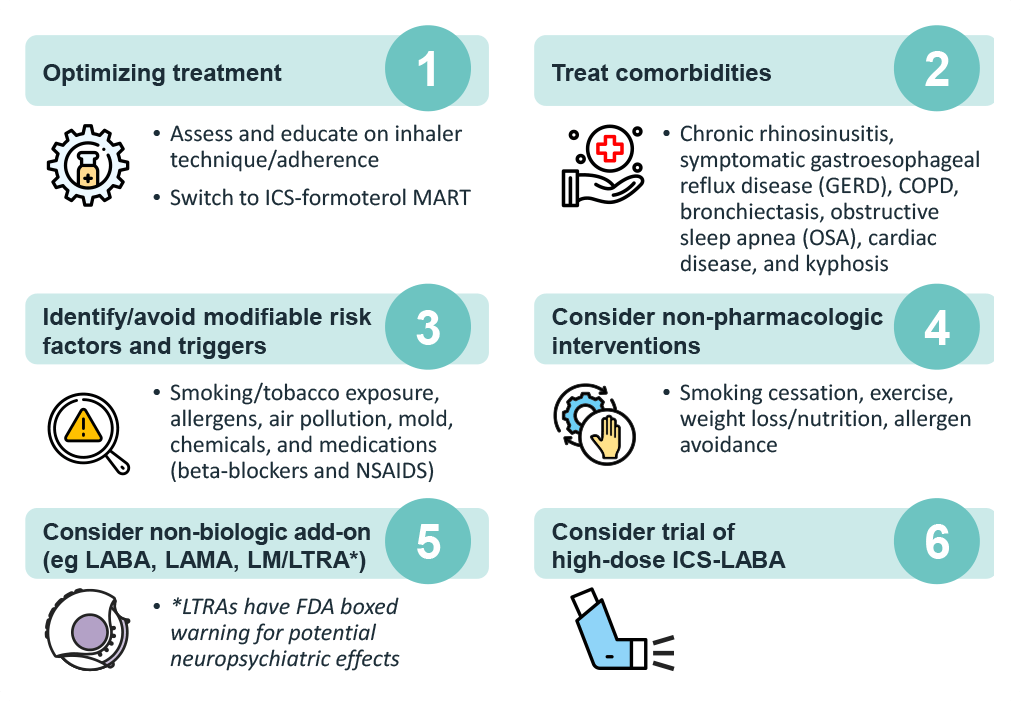
If asthma remains uncontrolled, referral to a specialist for assessment and management is indicated, preferably in a multidisciplinary severe asthma clinic (if available).3 It is also important to assess the asthma phenotype for evidence of type 2 inflammation.3 While many patients with mild-to-moderate asthma and type 2 inflammation rapidly improve when taking ICS consistently and correctly, severe asthma is refractory to high-dose ICS, leading to treatment with oral corticosteroids.3 Since OCS can suppress type 2 biomarkers, assessment for blood/sputum eosinophils and fractional exhaled nitric oxide [FeNO] should be performed before starting OCS, at least 1–2 weeks after an OCS course, or on the lowest possible dose of OCS.3 With evidence of type 2 inflammation, consider add-on type 2 targeted biologic therapy.3
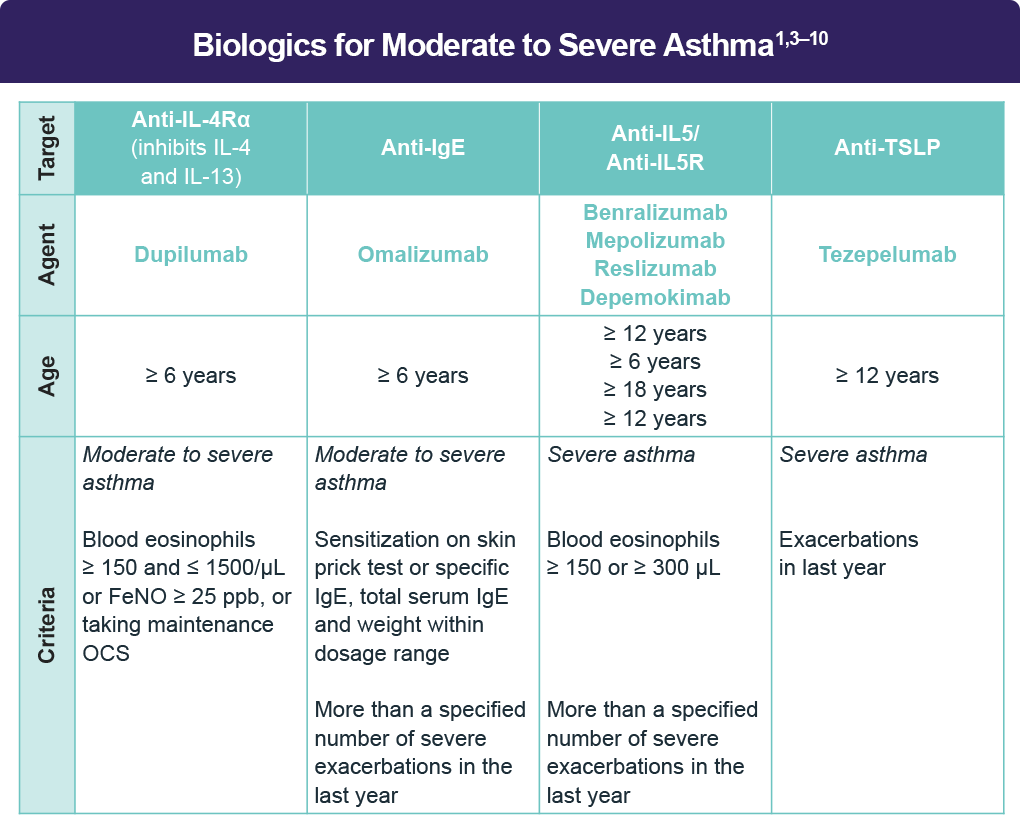
Clinical trial data for biologic therapy
Dupilumab4,11–13
The asthma clinical trials for patients aged 12 years and older included three randomized, double-blind, placebo-controlled, parallel-group, multicenter trials (DRI12544, QUEST, VENTURE). In DRI12544 (phase 2b), dupilumab was compared with placebo in adult subjects with moderate-to-severe asthma on a medium- or high-dose inhaled corticosteroid and a long-acting beta agonist. Significant increases in pre-bronchodilator FEV1 were observed at Week 12 for DRI12544 in the primary analysis populations (subjects with baseline blood eosinophil count of ≥ 300 cells/µL).4
In QUEST (phase 3), dupilumab was compared with placebo in 107 pediatric subjects 12 to 17 years of age and 1795 adult subjects with moderate-to-severe asthma on a medium- or high-dose ICS and a minimum of one and up to two additional controller medications. In the overall QUEST population, the rate of severe exacerbations was 0.46 and 0.52 for dupilumab 200 mg Q2W and 300 mg Q2W, respectively, compared to matched placebo rates of 0.87 and 0.97.4,11
VENTURE (phase 3) was a 24-week oral corticosteroid-reduction study in 210 adult and pediatric subjects 15 years of age and older with asthma who required daily oral corticosteroids in addition to regular use of high-dose inhaled corticosteroids plus an additional controller. Subjects continued to receive their existing asthma medicine during the study; however, their OCS dose was reduced every 4 weeks during the OCS reduction phase (Week 4–20), as long as asthma control was maintained. The mean percent reduction in daily OCS dose from baseline was 70% (median 100%) in subjects receiving dupilumab (95% CI, 60%–80%) compared to 42% (median 50%) in subjects receiving placebo (95% CI, 33%–51%).12
TRAVERSE, a single-arm, open-label extension study including participants from EXPEDITION (phase 2A), DRI12544 (phase 2b), QUEST (phase 3), and VENTURE (phase 3) evaluated participants aged 12 to 84 years with moderate to severe or OCS-dependent severe asthma up to 96 weeks. Study findings demonstrated sustained efficacy and safety with treatment extended up to 148 weeks, including rapid improvements in pre-bronchodilator FEV1, further improvements in annualized exacerbation rates, asthma control, and health-related quality of life, as well as progressively decreased serum eosinophils and total IgE.13
Omalizumab5
The safety and efficacy of omalizumab were evaluated in three randomized, double-blind, placebo-controlled, multicenter trials. All patients were required to have a baseline IgE between 30 and 700 IU/mL and body weight not more than 150 kg. Hospitalization rates were not significantly different between omalizumab and placebo-treated patients; however, the overall hospitalization rate was small. In asthma trials 1 and 2, all patients were symptomatic and were treated with ICS and short-acting beta2-agonists. After a 16-week run-in period with omalizumab, patients then entered an ICS reduction phase of 12 weeks during which ICS dose reduction was attempted in a step-wise manner. In both asthma trials 1 and 2, the number of exacerbations per patient was reduced in patients treated with omalizumab compared with placebo. In trial 3, there was no restriction on screening FEV1, and unlike trials 1 and 2, long-acting beta2-agonists were allowed. Patients were receiving at least 1000 mg/day fluticasone propionate, and a subset was also receiving oral corticosteroids. After a 16-week run-in period, patients entered an ICS reduction phase of 16 weeks during which ICS or oral steroid dose reduction was attempted in a step-wise manner. The number of exacerbations in patients treated with omalizumab was similar to that in placebo treated patients. The absence of an observed treatment effect may be related to differences in the patient population compared with trials 1 and 2, study sample size, or other factors.
Benralizumab6,14
In a 28-week randomized, controlled trial, researchers assessed the effects of benralizumab either every 4 weeks or every 8 weeks compared to placebo on the reduction in the oral glucocorticoid dose while asthma control was maintained in adult patients with severe asthma. The two benralizumab dosing regimens significantly reduced the median final oral glucocorticoid doses from baseline by 75%, as compared with a reduction of 25% in the oral glucocorticoid doses in the placebo group (P < .001 for both doses). The odds of an oral glucocorticoid dose reduction were more than four times as high with benralizumab than placebo. Among the secondary outcomes, benralizumab administered every 4 weeks resulted in an annual exacerbation rate that was 55% lower than the rate with placebo (P = .003), and benralizumab administered every 8 weeks resulted in an annual exacerbation rate that was 70% lower than the rate with placebo (P < .001). Frequencies of adverse events were similar between both benralizumab groups and the placebo group.
Mepolizumab7,15
In a randomized, double-blind trial in patients with severe eosinophilic asthma, the glucocorticoid-sparing effect of mepolizumab vs that of placebo was examined for 20 weeks. The primary outcome was the degree of reduction in the glucocorticoid dose (90 to 100% reduction, 75 to less than 90% reduction, 50 to less than 75% reduction, more than 0 to less than 50% reduction, or no decrease in oral glucocorticoid dose, a lack of asthma control during weeks 20 to 24, or withdrawal from treatment). The likelihood of a reduction in the glucocorticoid-dose stratum was 2.39 times greater in the mepolizumab arm than in the placebo arm (P = .008). The median percentage reduction from baseline in the glucocorticoid dose was 50% in the mepolizumab group, as compared with no reduction in the comparator (P = .007). Despite receiving a reduced glucocorticoid dose, patients in the mepolizumab arm, as compared with those in the placebo arm, had a relative reduction of 32% in the annualized rate of exacerbations (1.44 vs 2.12, P = .04) and a reduction of 0.52 points with respect to asthma symptoms, as measured on the Asthma Control Questionnaire 5.
Reslizumab8,16
Two duplicate, multicenter, double-blind, parallel-group, randomized, placebo-controlled, international, phase 3 trials enrolled patients aged 12–75 years, whose asthma was inadequately controlled by medium-to-high doses of inhaled corticosteroid-based therapy and who had blood eosinophils of 400 cells/μL or higher and one or more exacerbations in the previous year. Patients randomly received either intravenous reslizumab or placebo every 4 weeks for 1 year. The primary outcome was the annual frequency of clinical asthma exacerbations and was analyzed by intention to treat. In both studies, patients receiving reslizumab had a significant reduction in the frequency of asthma exacerbations (study 1: rate ratio 0.50; study 2: 0.41; both P < .0001) compared with those receiving placebo. Common adverse events on reslizumab were similar to placebo.
Tezepelumab9,17
Investigators conducted a phase 3, multicenter, randomized, double-blind, placebo-controlled trial (NAVIGATOR). Patients (12 to 80 years of age) were randomly assigned to receive tezepelumab or placebo subcutaneously for 52 weeks. The primary endpoint was the annualized rate of asthma exacerbations. This endpoint was assessed in patients with baseline blood eosinophil counts of <300 cells per microliter. Over 1000 patients underwent randomization with researchers reporting an annualized rate of asthma exacerbations of 0.93 (95% [CI] 0.80–1.07) with tezepelumab and 2.10 (95% CI, 1.84–2.39) with placebo (rate ratio, 0.44; P < .001).
In patients with a blood eosinophil count <300 cells per microliter, the annualized rate was 1.02 (95% CI, 0.84 to 1.23) with tezepelumab and 1.73 (95% CI, 1.46 to 2.05) with placebo (rate ratio, 0.59; P < .001). At week 52, improvements were greater with tezepelumab than with placebo relative to the prebronchodilator FEV1 (0.23 vs 0.09 liters; difference, 0.13 liters; P < .001).
Depemokimab10,18
Two phase 3, randomized, placebo-controlled replicate trials—SWIFT-1 and SWIFT-2— evaluated the efficacy and safety of depemokimab when administered in 6-month dosing intervals in patients (age ≥12 years) with severe, eosinophilic asthma. The primary endpoint was the annualized rate of exacerbations over 52 weeks. Pooled annualized exacerbation rates for 762 patients were significantly lower among those receiving depemokimab than those receiving placebo (0.51 and 1.11, respectively; P < .001). The probability that patients receiving depemokimab would have an exacerbation event over the 52-week trial was 32%. The proportion of patients with any adverse event was similar in both groups in both trials.
Treatment in pediatric populations (age 6–11 years)
Dupilumab19
In the 52-week EXCURSION study, children with moderate-to-severe asthma who previously participated in the VOYAGE study were enrolled to evaluate the long-term safety and efficacy of dupilumab. During EXCURSION, the safety profile and proportion of patients reporting treatment-emergent adverse events (TEAEs) were consistent with those observed during the parent study (VOYAGE). In the overall population, 232 (63.6%) of 365 patients experienced at least one TEAE (dupilumab/dupilumab: 147 [61.3%]; placebo/dupilumab: 85 [68.0%]). The most frequently reported TEAEs were nasopharyngitis, pharyngitis, and upper respiratory tract infections.
Omalizumab5,20
A 52-week study evaluated the safety and efficacy of omalizumab as add-on therapy in 628 pediatric patients aged 6 to <12 years with moderate-to-severe asthma inadequately controlled despite the use of ICS with or without other controller medications. The primary efficacy variable was the rate of asthma exacerbations during the 24-week, fixed steroid treatment phase. At 24 weeks, the omalizumab group had a statistically significantly lower rate of asthma exacerbations (0.45 vs 0.64) with an estimated rate ratio of 0.69 (95% CI, 0.53–0.90). The omalizumab group also had a lower rate of asthma exacerbations compared to placebo over the full 52-week double-blind treatment period (0.78 vs 1.36; rate ratio: 0.57; 95% CI, 0.45–0.72). The most common adverse reactions occurring at ≥3% in the pediatric patients receiving omalizumab and more frequently than in patients treated with placebo were nasopharyngitis, headache, pyrexia, upper abdominal pain, pharyngitis streptococcal, otitis media, viral gastroenteritis, arthropod bite, and epistaxis.
Mepolizumab7,21,22
A single open-label clinical trial (NCT02377427) was conducted in 36 children aged 6 to 11 years (mean age: 8.6 years, 31% female) with severe asthma over 12 weeks. The effectiveness of mepolizumab in pediatric patients was extrapolated from efficacy in adults and adolescents, with support from pharmacokinetic analyses showing similar drug exposure levels in pediatric vs adolescent and adult patients (40 mg subcutaneously every 4 weeks). The adverse reaction profile for patients aged 6 to 11 years was similar to that observed in patients aged 12 years and older. The most commonly reported manifestations of systemic allergic/hypersensitivity reactions in the group receiving mepolizumab included rash, pruritus, headache, and myalgia. The most commonly reported systemic non-allergic reactions in the group receiving mepolizumab included rash, flushing, and myalgia. In a long-term study extending over 52 weeks, all treatment groups experienced improved asthma control as well as reduced serum eosinophil counts and asthma exacerbations.
References
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. (2026 update). https://ginasthma.org/2026-gina-strategy-report/
- Beasley R, Bruce P, Houghton C, Hatter L. The ICS/formoterol reliever therapy regimen in asthma: A review. J Allergy Clin Immunol Pract. 2023;11:762-772.e1.
- Global Initiative for Asthma. Difficult-to-Treat & Severe Asthma in adolescent and adult patients. Diagnosis and Management. https://ginasthma.org/wp-content/uploads/2024/11/GINA-Severe-Asthma-Guide-2024-WEB-WMS.pdf
- Dupilumab (Dupixent®). Prescribing information. Regeneron Pharmaceuticals, Inc; 2025. https://www.regeneron.com/downloads/dupixent_fpi.pdf
- Omalizumab (Xolair®). Prescribing information. Genentech, Inc; 2024. https://www.gene.com/download/pdf/xolair_prescribing.pdf
- Benralizumab (Fasenra®) Prescribing information. AstraZeneca; 2024. https://www.azpicentral.com/pi.html?product=fasenra
- Mepolizumab (Nucala®) Prescribing information. GSK; 2025. https://gskpro.com/content/dam/global/hcpportal/en_US/Prescribing_Information/Nucala/pdf/NUCALA-PI-PIL-IFU-COMBINED.PDF
- Reslizumab (Cinqair®) Prescribing information. Teva Respiratory, LLC; 2020. https://www.cinqair.com/globalassets/cinqair/prescribinginformation.pdf
- Tezepelumab-ekko (Tezpire®) Prescribing information. AstraZeneca; 2023. https://www.azpicentral.com/pi.html?product=tezspire
Depemokimab (Exdensur®) Prescribing information. GSK;2025. https://gskpro.com/content/dam/global/hcpportal/en_US/Prescribing_Information/Exdensur/pdf/EXDENSUR-PI-PIL.PDF
- Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med. 2018;378:2486-2496.
- Ribas CD, Maspero J, Castro M, et al. Dupilumab reduced oral corticosteroid use and improved clinical outcomes regardless of baseline OCS dose in patients with uncontrolled, severe asthma in the liberty asthma VENTURE study. Chest. 2021;160 (4 suppl):1893A-1897A.
- Wechsler ME, Ford LB, Maspero JF, et al. Long-term safety and efficacy of dupilumab in patients with moderate-to-severe asthma (TRAVERSE): An open-label extension study. Lancet Respir Med. 2022;10:11-25.
- Nair P, Wenzel S, Rabe KF, et al. Oral glucocorticoid–sparing effect of benralizumab in severe asthma. N Engl J Med. 2017;376:2448-2458.
- Bel EH, Wenzel SE, Thompson PJ, et al. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371:1189-1197.
- Castro M, Zangrilli J, Wechsler ME, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: Results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015;3:355-366.
- Menzies-Gow A, Corren J, Bourdin A, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. 2021;384:1800-1809.
- Jackson DJ, Wechsler ME, Jackson DJ, et al. Twice-yearly depemokimab in severe asthma with an eosinophilic phenotype. N Engl J Med. 2024;391:2337-2349.
- Bacharier LB, Maspero JF, Katelaris CH, et al. Assessment of long-term safety and efficacy of dupilumab in children with asthma (LIBERTY ASTHMA EXCURSION): An open-label extension study. Lancet Respir Med. 2024;12:45-54.
- Lanier B, Bridges T, Kulus M, Taylor AF, Berhane I, Vidaurre CF. Omalizumab for the treatment of exacerbations in children with inadequately controlled allergic (IgE-mediated) asthma. J Allergy Clin Immunol. 2009;124:1210-1216.
- Gupta A, Pouliquen I, Austin D, et al. Subcutaneous mepolizumab in children aged 6 to 11 years with severe eosinophilic asthma. Pediatr Pulmonol. 2019;54:1957-1967.
- Gupta A, Ikeda M, Geng B, et al. Long-term safety and pharmacodynamics of mepolizumab in children with severe asthma with an eosinophilic phenotype. J Allergy Clin Immunol. 2019;144:1336-1342.e7.
All URLs accessed May 7, 2026
