Pathophysiology
Pathophysiology of Asthma
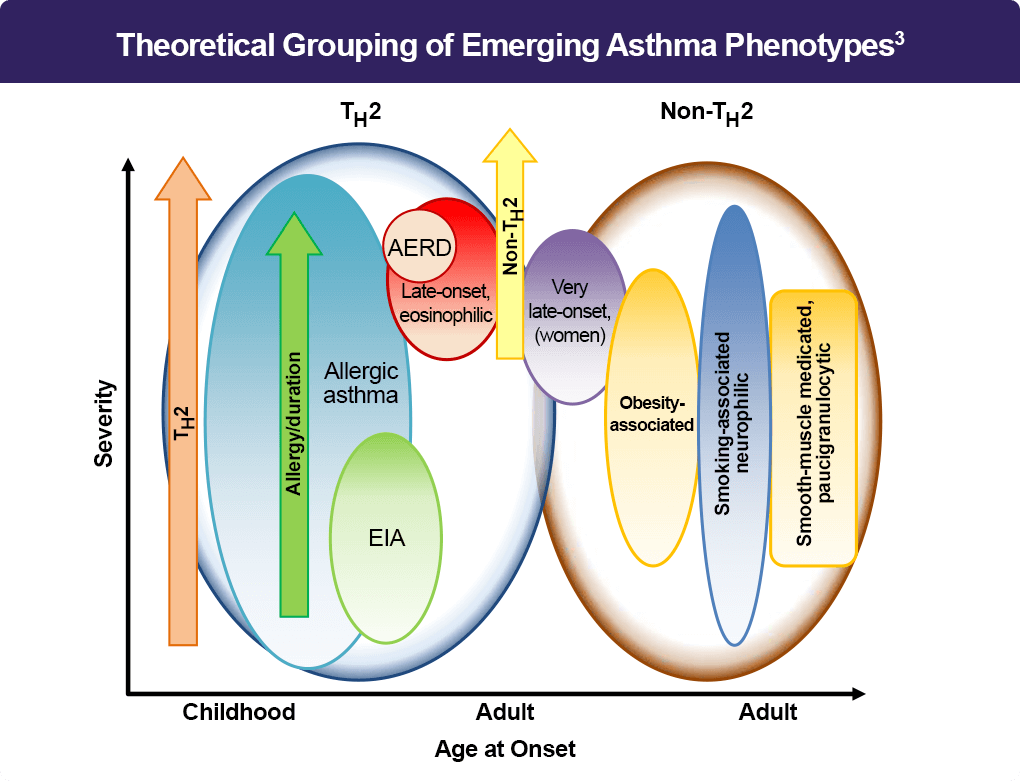
Asthma is a heterogeneous lung disease with variable phenotypes (clinical presentations) and distinctive endotypes (mechanisms). Aberrant T helper type 2 (Th2) inflammation is the most important pathological process for asthma, which is mediated by Th2 cytokines, such as interleukin (IL)-5, IL-4, and IL-13. Approximately 50% of mild to moderate asthma and a large portion of severe asthma are induced by Th2-dependent inflammation.1 Consequently, the distinction between Th2-mediated and non-Th2 disease applies across all levels of severity. Recently, based on the status of Th2 inflammation, the disease has been classified into two groups: Th2-high (mediated) and Th2-low asthma.2 Th2-high asthma is characterized by eosinophilic airway inflammation, which is associated with increased blood eosinophil counts or elevations of fractional exhaled nitric oxide (FeNO).2,3 Whereas Th2-low asthma includes neutrophilic asthma and paucigranulocytic asthma.3 The coexistence of eosinophilic and neutrophilic airway inflammation is considered mixed granulocytic asthma.3
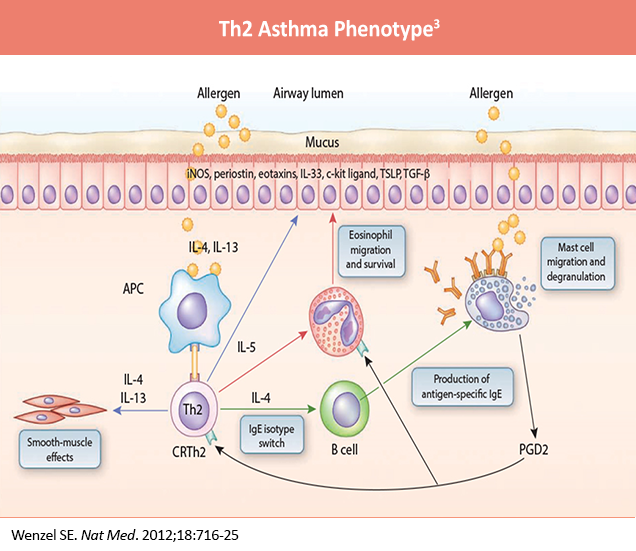
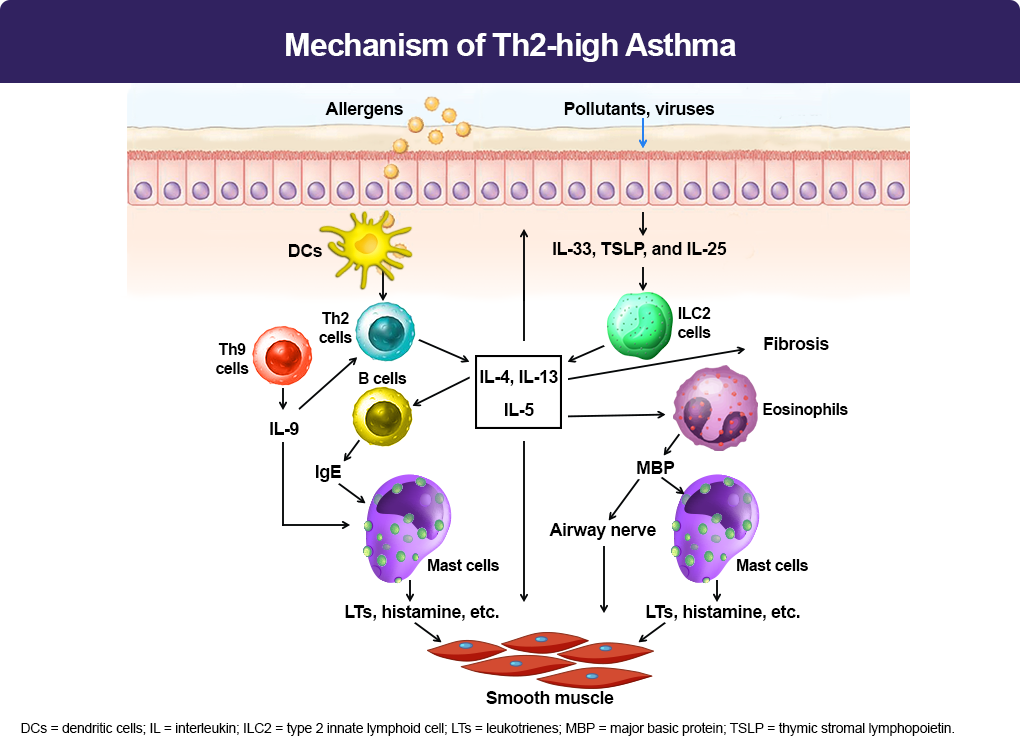
When allergens enter the lower airways, dendritic cells (DCs) present the allergens to Th2 cells, which secrete Th2 cytokines, including interleukin (IL)-5, IL-4, and IL-13. IL-4 and IL-13 activate B cells, which produce IgE.3 IgE subsequently binds to the surface of mast cells. When the same allergens enter the airways, they interact with IgE, which induces mast cells to release mediators, such as leukotrienes (LTs), histamine, and ILs. These mediators irritate airway smooth muscle and induce bronchoconstriction. In addition, IL-5 facilitates eosinophil recruitment to the lungs. Eosinophils also release mediators, including major basic protein (MBP), which stimulates mast cells to release histamines and LTs. MBP also inhibits M2 receptors, promotes acetylcholine release from cholinergic nerves, and induces bronchospasm.3 Furthermore, IL-13 directly sensitizes airway smooth muscle contraction, stimulates epithelial cells to secrete mucins, and induces fibrosis. Th9 cells can secrete IL-9, which activates Th2 cells and promotes mast cell accumulation. Lastly, epithelium injury by infection and pollutants induces release of cytokines, including thymic stromal lymphopoietin (TSLP), IL-25, and IL-33, which activate type 2 innate lymphoid cells (ILC2) and produce Th2 cytokines, such as IL-5 and IL-13.3
References
- Brusselle GG, Koppelman GH. Biologic therapies for severe asthma. N Engl J Med. 2022;386:157-171.
- Pelaia C, Melhorn J, Hinks TS, et al. Type 2 severe asthma: pathophysiology and treatment with biologics. Expert Rev Respir Med. 2024;18(7):485-498.
- Habib N, et al. Current understanding of asthma pathogenesis and biomarkers. Cells. 2022;11:2764.
